


IF:11.4,网络药理学+机器学习+铁死亡研究红景天苷使三阴性乳腺癌铁死亡的机制!!!
题目:红景天苷通过SCD1介导的脂肪生成和NCOA4介导的铁蛋白自噬使三阴性乳腺癌对铁死亡敏感
英文名:Salidroside sensitizes Triple-negative breast cancer to ferroptosis by SCD1-mediated lipogenesis and NCOA4-mediated ferritinophagy
杂志:Journal of Advanced Research
影响因子:11.4
发表时间:2024年9月29日
研究背景:阴性乳腺癌(TNBC)是女性乳腺癌死亡的主要原因。文献已证实红景天苷(Sal)在治疗TNBC中的益处。然而,关于Sal治疗TNBC的潜在治疗靶点和机制的研究仍然有限。本研究旨在探讨Sal抗TNBC的主要靶点和潜在机制。
研究思路:整合网络药理学、生物信息学和机器学习算法策略,以研究Sal在TNBC中的作用、潜在靶点和机制。选用MDA-MB-231细胞和荷瘤裸鼠进行体外和体内实验。采用CCK-8、LDH检测和钙黄绿素-AM/PI染色测定细胞活力和细胞毒性。使用谷胱甘肽、谷胱甘肽过氧化物酶、丙二醛(MDA)、C11-BODIPY581/591探针和FerroOrange染料检测抗氧化防御、脂质过氧化和铁代谢。通过过表达谷胱甘肽过氧化物酶4(GPX4)或硬脂酰辅酶A去饱和酶1(SCD1)以及敲低核受体共激活因子4(NCOA4)来论证Sal对TNBC的作用机制。预测结果证实,在Sal和TNBC中鉴定出22个铁死亡相关基因,表明Sal作用于TNBC的潜在机制与铁死亡相关。此外,功能富集分析显示这些基因主要参与mTOR、PI3K/AKT和自噬信号通路。体外验证结果表明,Sal通过调节铁死亡抑制TNBC细胞增殖,表现为细胞内Fe2+水平升高和脂质过氧化。机制上,Sal通过抑制PI3K/AKT/mTOR轴使TNBC细胞对铁死亡敏感,从而抑制SCD1介导的单不饱和脂肪酸脂肪生成以诱导脂质过氧化,同时促进NCOA4介导的铁蛋白自噬以增加细胞内Fe2+含量。GPX4或SCD1过表达或NCOA4缺失的结果进一步支持了我们的机制研究。体内实验证实,Sal通过诱导铁死亡对减缓肿瘤生长至关重要。
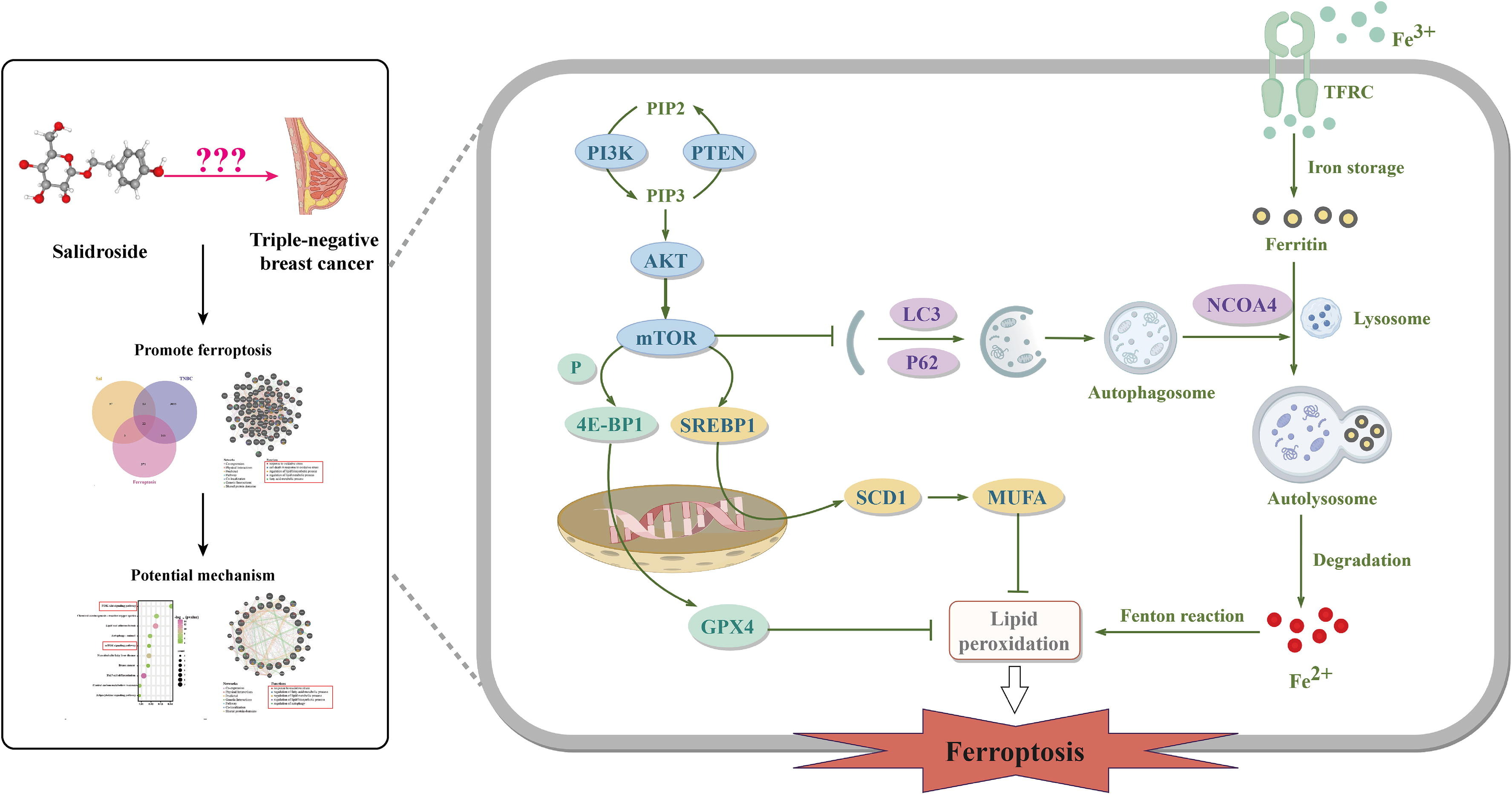
图1
研究结果:
1、Sal和三阴性乳腺癌(TNBC)相关靶点集合
为了获得红景天苷(Sal)的作用靶点,以“红景天苷”为关键词搜索药物相关数据库。结果发现,排除重复项后,共剩下178个靶点(图2A)。随后,通过条件筛选获得了1374个三阴性乳腺癌(TNBC)相关靶点(图2B)。
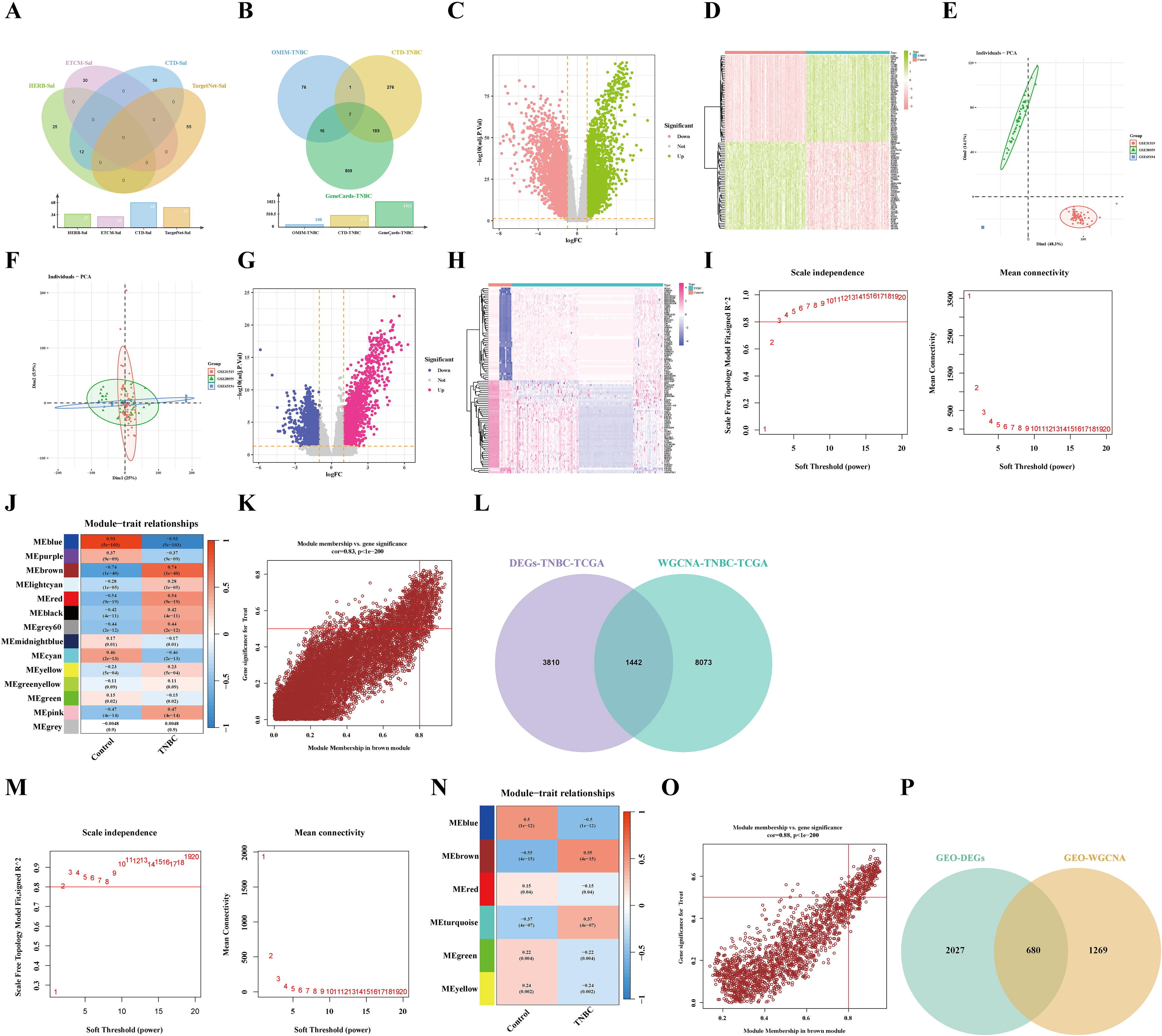
图2
2、三阴性乳腺癌中差异表达基因及关键模块基因的鉴定
为了提高结果的可靠性,从TCGA和GEO数据集下载了三阴性乳腺癌(TNBC)样本进行差异表达基因(DEG)分析。结果发现,在TCGA样本中识别出5252个差异表达基因,其中2666个下调的差异表达基因和2586个上调的差异表达基因(图2C-D)。同时,在GEO数据集中进行了差异表达基因分析。然后为所有数据集生成主成分分析(PCA)图(图2E-F)。此外,确定了2707个差异表达基因,其中包括1340个下调的差异表达基因和1367个上调的差异表达基因(图2G-H)。
为了识别与三阴性乳腺癌(TNBC)相关的关键模块,对TCGA-TNBC样本进行了加权基因共表达网络分析(WGCNA)(图2I)。此外,确定了14个模块,其中棕色模块与三阴性乳腺癌高度相关(图2J)。此外,散点图显示棕色模块与三阴性乳腺癌之间存在显著关系(cor=0.83, p < 1e − 200)(图2K)。因此,通过将WGCNA的棕色模块相关基因与Limma鉴定的差异表达基因相交,获得了1442个基因(图2L)。GEO数据集显示软阈值幂为3,对应未缩放拓扑拟合指数达到0.88(图2M)。确定了六个模块,棕色模块与三阴性乳腺癌高度相关(图2N)。此外,散点图显示蓝色模块与三阴性乳腺癌之间存在正显著关系(cor=0.88,p<1e−200)(图2O)。因此,通过将WGCNA的棕色模块相关基因与Limma鉴定的差异表达基因相交,获得了680个基因(图2P)。
3、预测和实验分析结果阐明Sal通过促进铁死亡抗三阴性乳腺癌
此外,通过疾病、TCGA和GEO数据库的交集获得了3296个三阴性乳腺癌相关基因(图3A)。Sal基因与三阴性乳腺癌基因的交集获得了75个共有基因(图3B)。为了探索Sal对三阴性乳腺癌的潜在作用机制,利用GeneMANIA工具对75个共有基因进行功能分析。结果显示,共有基因主要参与与氧化应激、氧化应激反应、氧化应激下的细胞死亡、脂质生物合成过程的调节以及脂质代谢过程的调节等过程(图3C)。铁死亡是一种由脂质过氧化和氧化还原失衡驱动的铁依赖性程序性细胞死亡形式[36]。在引起细胞氧化应激的因素中,膜双层中脂质的氧化修饰,特别是脂质过氧化,已成为细胞命运的重要调节因子。广泛的脂质过氧化通过一种称为铁死亡的独特细胞死亡机制使细胞死亡[37]。因此,我们推测Sal抗三阴性乳腺癌的潜在机制可能与铁死亡有关。
最初,我们在MDA-MB-231细胞中验证了Sal对三阴性乳腺癌(TNBC)的作用。观察到Sal抑制细胞活性与治疗的剂量和时间相关(补充图S1)。基于此,选择了Sal的IC50进行进一步研究。此外,铁死亡诱导剂Erastin可以抑制细胞活性与治疗剂量相关(图3D)。相反,这种作用可以被Fer-1逆转(图3E),表明MDA-MB-231细胞中发生了铁死亡。我们进一步验证了Sal是否通过触发铁死亡改善三阴性乳腺癌。将铁死亡抑制剂Fer-1、DFO和GSH添加到MDA-MB-231细胞中。结果显示,通过使用Fer-1、DFO和GSH等铁死亡抑制剂处理,Sal对细胞活性的抑制作用部分得到缓解(图3F-G)。在后续实验中选择Fer-1进行进一步研究,它主要通过调节细胞内抗氧化系统和铁代谢来抑制铁死亡。钙黄绿素-AM/PI染色显示,通过给予Fer-1处理,细胞活性抑制部分得到缓解(图3H)。随后,添加Fer-1显著提高了谷胱甘肽(GSH)水平。相比之下,Sal降低了谷胱甘肽过氧化物酶(GPX)活性(图3I-J),丙二醛(MDA)、Fe2+和脂质活性氧(ROS)水平在Sal组中均显著增加(图3K-M)。然而,Sal组中的上述结果部分被Sal+Fer-1逆转,表明Sal在MDA-MB-231细胞中诱导铁死亡。
同时,对于Sal在体内的潜在抗癌活性,通过在雌性裸鼠中接种MDA-MB-231细胞获得了三阴性乳腺癌动物模型(图3N)。如图所示,与单独使用Fer-1和联合给药组相比,Sal组的肿瘤生长明显较慢。Fer-1单独给药组和联合给药组(图3O-P)。此外,Sal组的肿瘤重量显著低于其他组(图3Q)。在治疗期间,体重也只有很小的变化,表明这些治疗没有明显的毒性,并且耐受性良好(图3R)。此外,肿瘤组织的HE染色结果显示,Sal治疗组的坏死区域比其他组大得多(图3S)。
此外,在施用Sal和Fer-1后,Ki67阳性区域的上调比仅接受Sal的区域更为明显(图3S)。所有数据表明,Sal可以对抗三阴性乳腺癌而没有明显的毒性作用。随后,与其他组相比,Sal组的谷胱甘肽和谷胱甘肽过氧化物酶活性水平显著降低,但丙二醛和Fe2+水平升高(图3T-W)。然而,这一结果与联合组的结果相反。此外,免疫组化结果发现,Sal组中的4-HNE增强,表明Sal促进脂质过氧化物的积累(图3X)。同时,在免疫组化分析中,Sal组中PTGS2的阳性表达升高。这支持了Sal通过诱导铁死亡在抑制三阴性乳腺癌肿瘤生长中起关键作用的观点(图3X)。
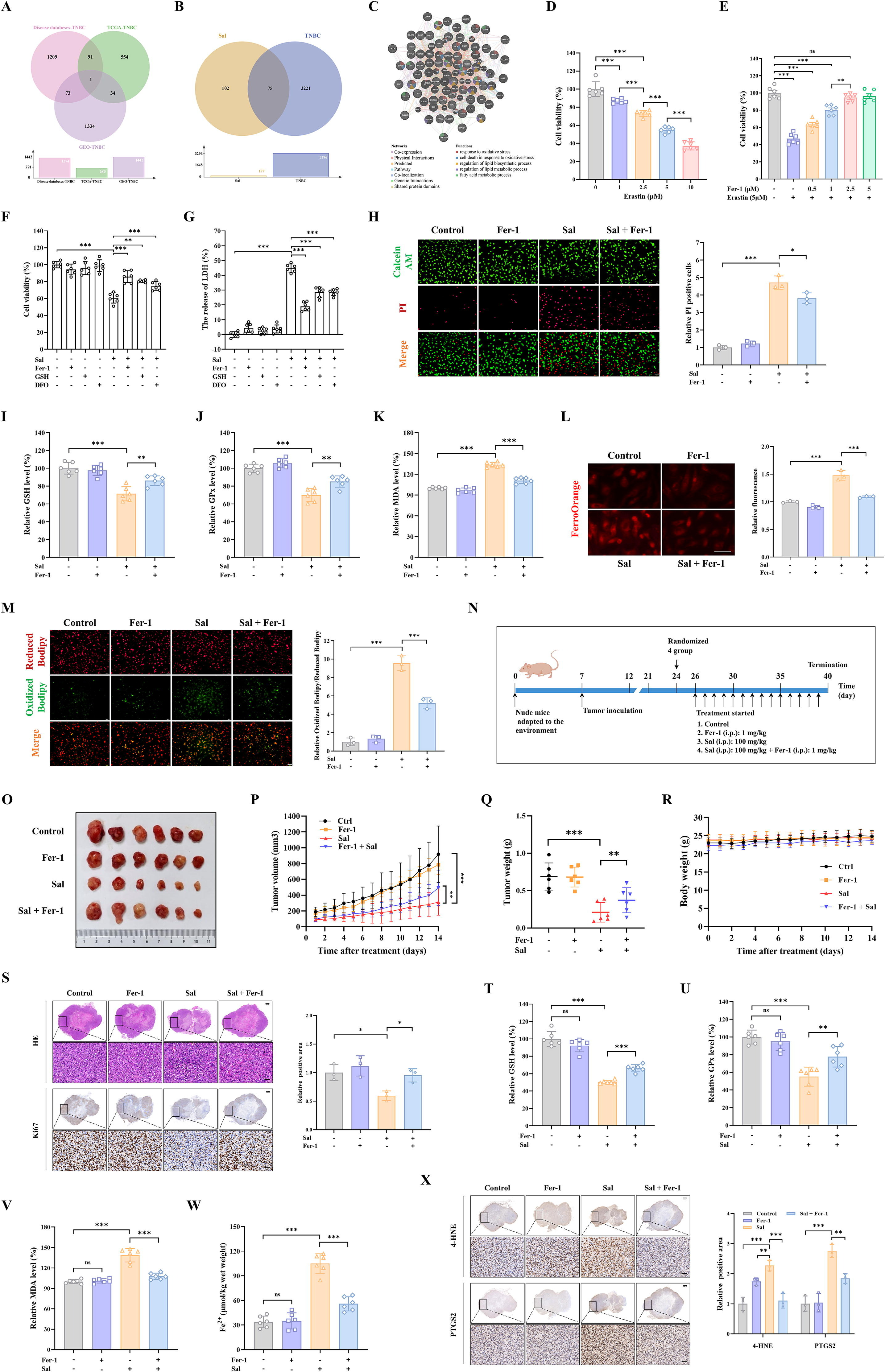

图3
4、共享FRGs的功能富集分析结果
在Sal和三阴性乳腺癌(TNBC)中鉴定出22个与铁死亡相关的基因,这些基因被定义为共享的铁死亡相关基因(FRGs)(图4A)。并进行了GO分析显示(图4B-C)。
此外,Sal治疗三阴性乳腺癌(TNBC)通过触发铁死亡的功效与KEGG富集通路紧密相关,即脂质和动脉粥样硬化(hsa05417)、mTOR信号通路(hsa04150)、PI3K/AKT信号通路(hsa04151)和自噬-动物(hsa04140)信号通路(图4D)。这些基因与氧化应激、脂质和脂肪酸代谢过程、脂质生物合成过程以及自噬过程的调节密切相关(图4E)。
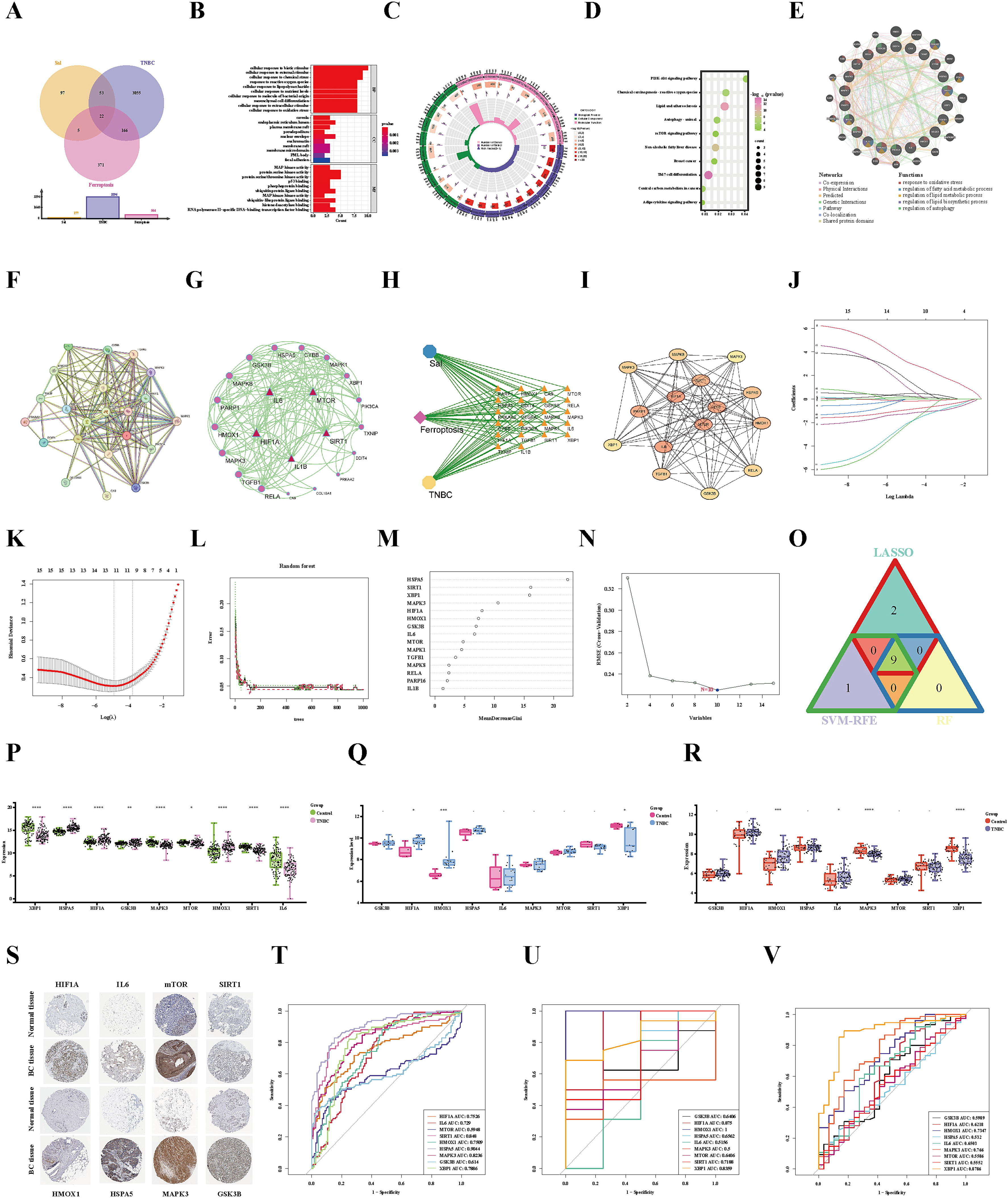
图4
5、PPI网络构建及关键FRGs鉴定
为了研究常见FRGs处的蛋白质相互作用,使用STRING在线工具构建了一个蛋白质相互作用(PPI)网络(图4F-G)。同时,构建了一个药理网络来表示Sal-FRGs-TNBC之间的关系(图4H)。使用Cytohubba插件计算了前15个中心基因(图4I)。
6、共享FRGs中筛选枢纽特征基因
本研究结合三种机器学习算法,从PPI网络筛选出的15个枢纽基因中识别出与Sal和TNBC相关的潜在诊断生物标志物。首先,通过LASSO算法获得了11个生物标志物(图4J-K)。同时,RF算法根据重要性确定了九个潜在生物标志物(图4L-M)。此外,SVM-RFE分析表明,涉及十个基因(HSPA5、SIRT1、XBP1、MAPK3、GSK3B、HMOX1、HIF1A、IL6、MAPK1和mTOR)的模型实现了highest的准确性(图4N)。最后,三种机器学习算法交叉识别基因,以获得候选生物标志物,即HIF1A、IL6、mTOR、SIRT1、HMOX1、HSPA5、MAPK3、GSK3B和XBP1(图4O)。
为了进一步研究候选生物标志物在三阴性乳腺癌(TNBC)患者中的重要性,分析了上述基因在TNBC患者和正常样本中的表达情况。在TCGA-TNBC测试集中,HIF1A、mTOR、HMOX1、HSPA5和GSK3B的表达水平在TNBC样本中显著升高,而IL6、SIRT1、MAPK3和XBP1的表达降低。这些差异均具有统计学意义(图4P)。同时,验证数据集GSE61724与上述结果一致,除MAPK3外,所有基因均表现出一致的趋势(图4Q)。此外,验证数据集GSE21653的结果发现,除了SIRT1、MAPK3和XBP1的表达降低外,其余基因在TNBC患者中均有表达(图4R)。此外,使用人类蛋白质图谱(HPA)数据库中的免疫组化结果验证了上述基因的表达。免疫组化结果与上述结果一致。除XBP1外,HPA数据库中没有数据。HIF1A、IL6、mTOR、SIRT1、HMOX1、HSPA5、MAPK3和GSK3B在乳腺癌组织中表现出相同的增加趋势(图4S)。在TCGA-TNBC数据集中,所有基因的AUC值均≥0.6,表明具有极好的诊断价值(图4T)。同时,在GSE61724和GSE21653验证集中获得的结果与上述结果几乎一致。在验证集中,所列基因的平均AUC值均≥0.6,除了IL6和HSPA5(图4U-V)。这些结果为HIF1A、mTOR、SIRT1、HMOX1、GSK3B、MAPK3和XBP1作为三阴性乳腺癌的诊断生物标志物提供了有力证据。
7、Sal通过调节MDA-MB-231细胞和三阴性乳腺癌动物模型中的PI3K/AKT/mTOR信号通路诱导铁死亡
在体外实验中,结果显示磷酸酶和张力蛋白同源物(PTEN)的蛋白表达水平增加。然而,在Sal组中,p-PI3K/PI3K和p-AKT/AKT降低(图5a),表明Sal触发的铁死亡可能抑制PI3K/AKT通路。为了进一步验证Sal诱导铁死亡的机制,应用PTEN抑制剂VO-Ohpic三水合物和AKT激活剂SC79,以探索PTEN和AKT在PI3K/AKT通路调节的铁死亡中的重要作用。结果表明,VO-Ohpic三水合物和SC79使MDA-MB-231细胞对Sal诱导的铁死亡具有抗性(图5B-C)。除此之外,VO-Ohpic三水合物和SC79还显著降低了Sal诱导的丙二醛(MDA)并提高了亚铁水平(图5D-E)。PI3K只是调节AKT激活的众多机制之一,此外,VO-Ohpic三水合物可以提高Sal触发的p-PI3K/PI3K和p-AKT/AKT(图5F)。
为了进一步验证预测结果,通过WB检测了枢纽生物标志物mTOR的表达水平。给予Sal后,p-mTOR/mTOR蛋白显著下调(图5G)。此外,作为mTORC1的底物,p4EBP1/4EBP1在蛋白水平上给予Sal后下调(图5G)。之后,应用mTORC1激活剂MHY1485和抑制剂雷帕霉素进一步探索mTORC1在Sal调节的铁死亡中的作用。结果表明,MHY1485预处理赋予细胞对Sal引起的铁死亡的抗性,而雷帕霉素增强了Sal诱导的铁死亡(图5H-I)。此外,MHY1485大大下调了Sal调节的MDA生成,而雷帕霉素则增加了(图5J)。Sal诱导的PTGS2蛋白水平升高被MHY1485显著抑制(图5K)。上述发现与预测结果一致。PI3K/AKT/mTOR的活性可能与MDA-MB-231细胞对Sal诱导的铁死亡敏感性增加有关。
值得注意的是,铁死亡PI3K/AKT/mTOR信号通路在体内得到了验证。在给予Sal后,PTEN的蛋白水平上调,而p-PI3K/PI3K、p-AKT/AKT和p-mTOR/mTOR下调(图5L)。总之,这些结果表明,Sal可能通过调节与mTOR相关的PI3K/AKT/mTOR通路诱导铁死亡来改善三阴性乳腺癌。
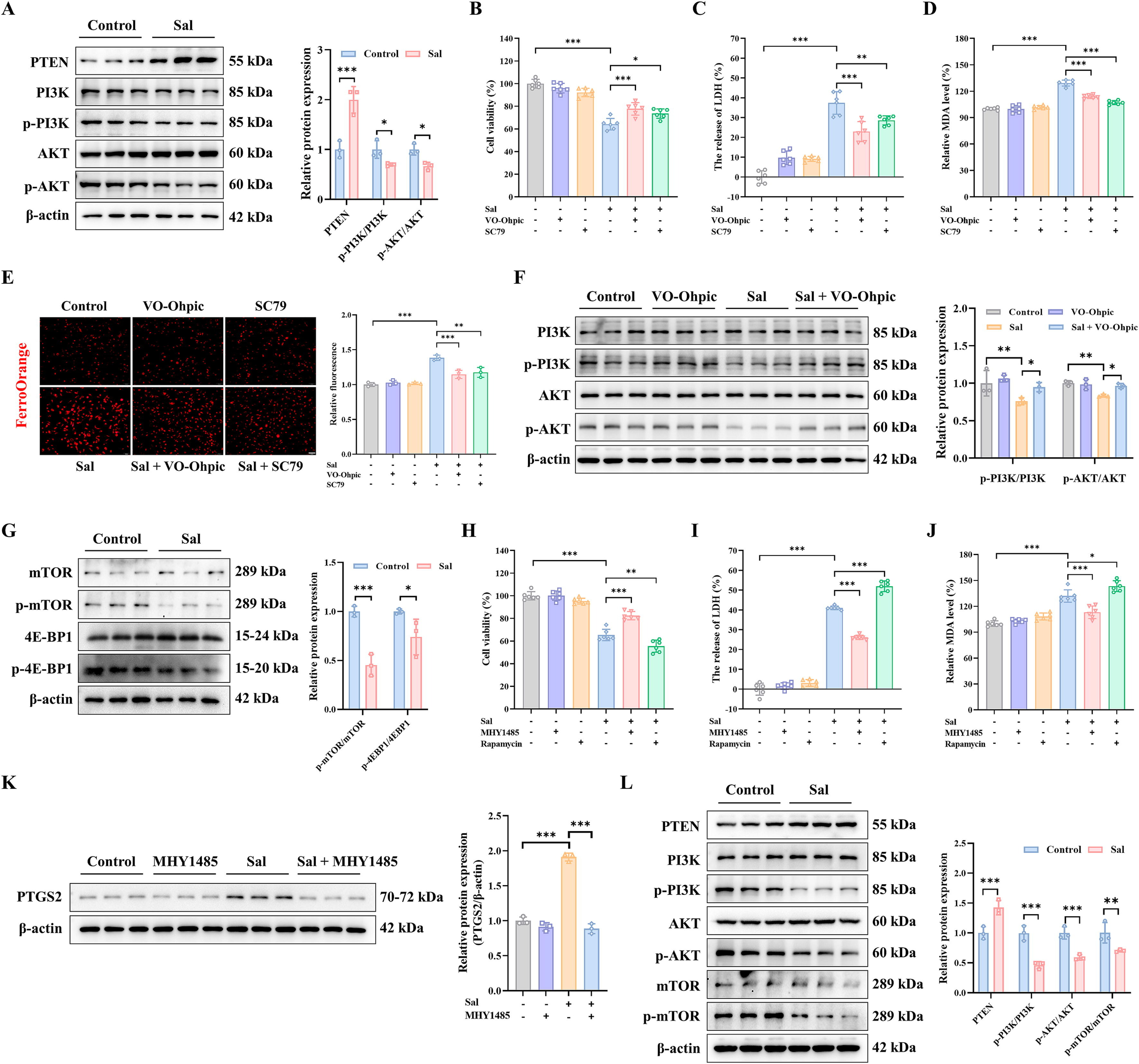
图5
8、Sal通过调节MDA-MB-231细胞和三阴性乳腺癌动物模型中mTOR介导的GPX4抗氧化防御诱导铁死亡
通过RT-PCR和WB方法测量GPX4和PTGS2以测试预测结果。结果发现Sal可以显著降低GPX4水平,而PTGS2在mRNA和蛋白质水平上增加(图6A-B)。一致地,mTOR激活剂MHY1485也显著改善了Sal诱导的GPX4表达抑制(图6C)。为了进一步研究GPX4是否与Sal调节的铁死亡有关,在MDA-MB-231细胞中获得了GPX4过表达(图6D)。与Fer-1给药一致,通过Sal处理,GPX4过表达的MDA-MB-231细胞活性抑制得到部分缓解(图6E,H)。通常,MDA和PTGS2水平显著下调(图6G,I),而由于GPX4过表达,GPX活性增加(图6F)。因此,Sal诱导的铁死亡可能归因于GPX4的降低。此外,数据证实了在Sal处理的MDA-MB-231细胞中发生了铁死亡,并且Sal触发铁死亡的机制可能是通过GPX4信号通路调节mTOR介导的脂质过氧化。
此外,在三阴性乳腺癌动物模型中测试了GPX4信号通路在抗氧化防御过程中的作用。结果与体外实验一致。GPX4的mRNA和蛋白质水平降低。相反,Sal处理后PTGS2水平升高(图6J-K),这表明Sal可以治疗三阴性乳腺癌。其机制可能是通过抑制脂质过氧化中mTOR介导的GPX4信号通路来诱导铁死亡。
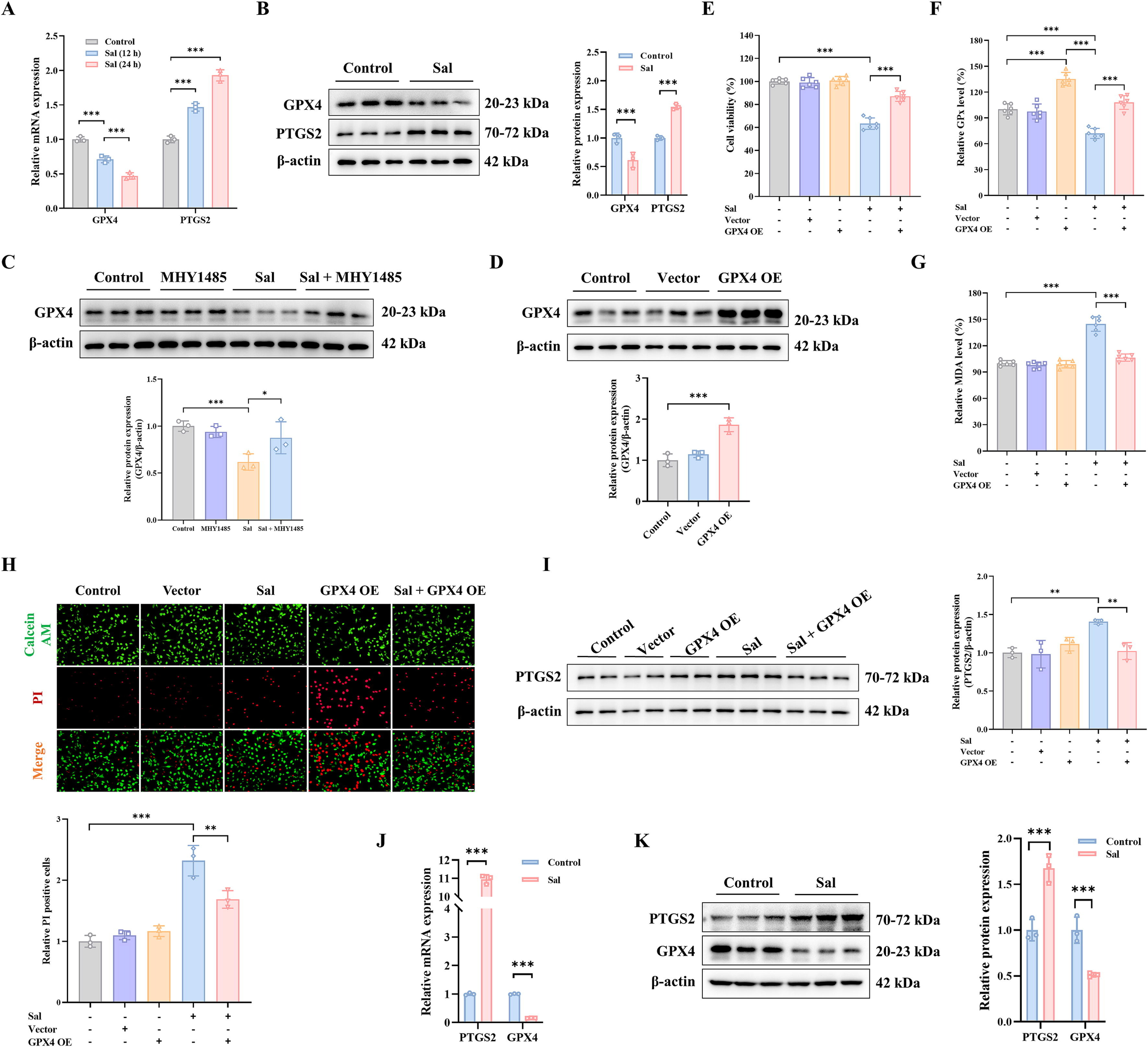
图6
9、Sal通过抑制脂肪酸合成中mTOR介导的SCD1通路在MDA-MB-231细胞和TNBC动物模型中诱导铁死亡
Sal处理显著降低了甾醇调节元件结合蛋白1(SREBP1)的mRNA和蛋白质水平(图7A-B)。此外,由于SREBP1影响与脂质合成相关的基因转录,因此检测了下游相关基因的mRNA,包括乙酰辅酶A羧化酶(ACACA)、ATP柠檬酸裂解酶(ACLY)、脂肪酸合酶(FASN)和硬脂酰辅酶A去饱和酶(SCD)。相比之下,SCD显著下调(图7A)。同时,Sal处理时SCD1的蛋白质表达显著降低(图7B)。此外,mTOR激活剂MHY1485增加了Sal诱导的SREBP1和SCD1的蛋白质表达(图7C)。SCD1可以调节单不饱和脂肪酸(MUFAs)的生成,与饱和脂肪酸(SFAs)的转化有关。因此,MDA-MB-231细胞预先用单不饱和脂肪酸OA(18:1)和饱和脂肪酸SA(18:0)处理。最后,由于是OA而不是SA,MDA-MB-231细胞似乎在更大程度上对Sal诱导的铁死亡具有抗性(图7D)。通过降低Sal诱导的MDA水平也证实了这种抑制作用(图7E)。总体而言,研究结果表明,Sal抑制了由SCD1产生的单不饱和脂肪酸OA的合成,这是SCD1诱导铁死亡的主要原因。
为了证明Sal诱导的铁死亡归因于硬脂酰辅酶A去饱和酶1(SCD1),在MDA-MB-231细胞中构建了插入SCD1的过表达质粒(图7F)。我们发现过表达的SCD1可以增加Sal诱导的细胞活力(图7G-H)。同时,在SCD1过表达时,丙二醛(MDA)和脂质活性氧(ROS)水平显著降低(图7I-J)。值得注意的是,过表达的SCD1也抑制了Sal诱导的谷胱甘肽过氧化物酶4(GPX4)和前列腺素内过氧化物合酶2(PTGS2)的蛋白表达(图7K)。这些发现共同表明,Sal可能通过抑制脂肪酸合成中mTOR介导的SCD1信号通路促进MDA-MB-231细胞的铁死亡。

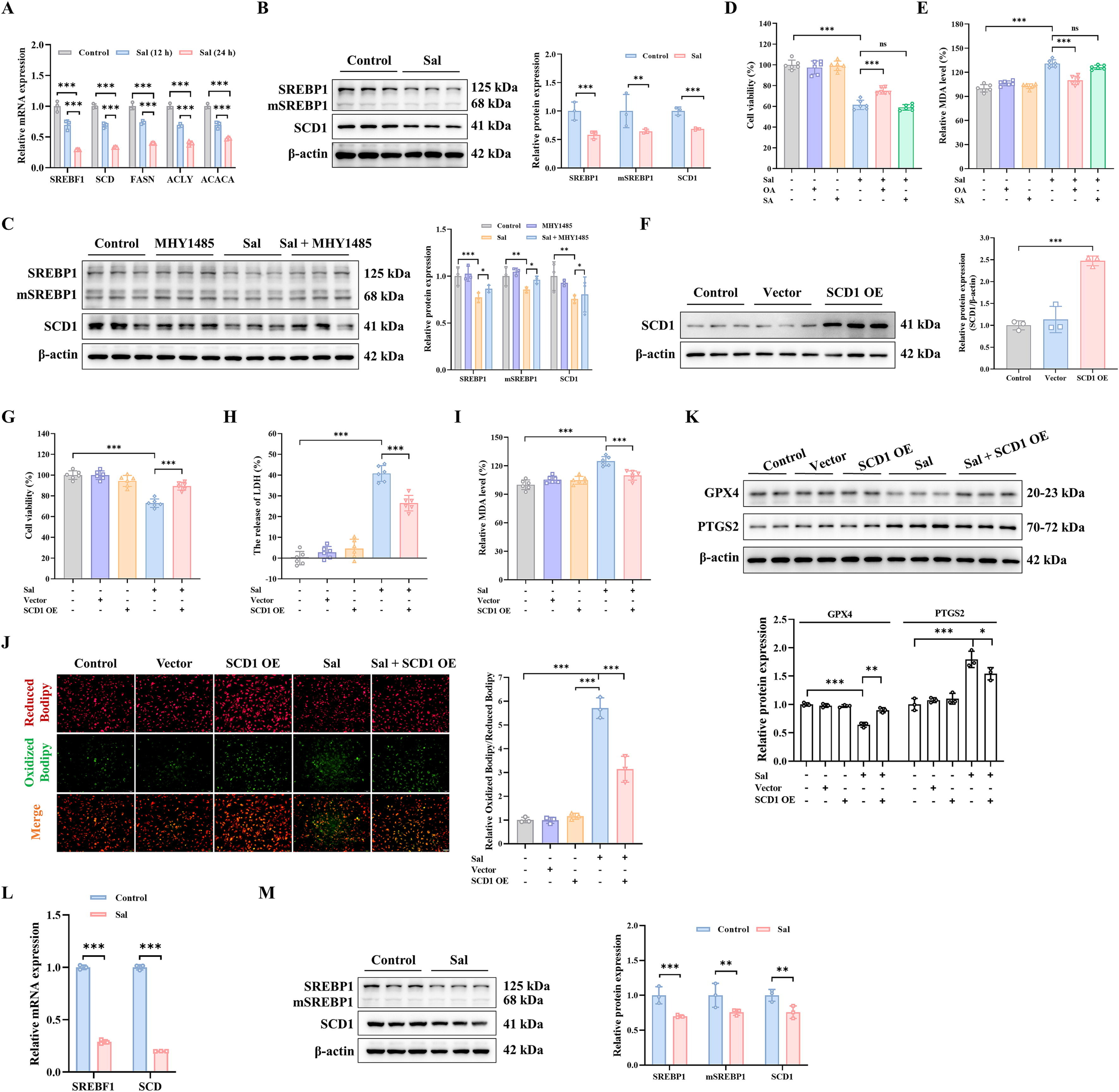
图7
10、Sal在MDA-MB-231细胞和三阴性乳腺癌动物模型中促进由mTOR调节的NCOA4介导的铁蛋白自噬
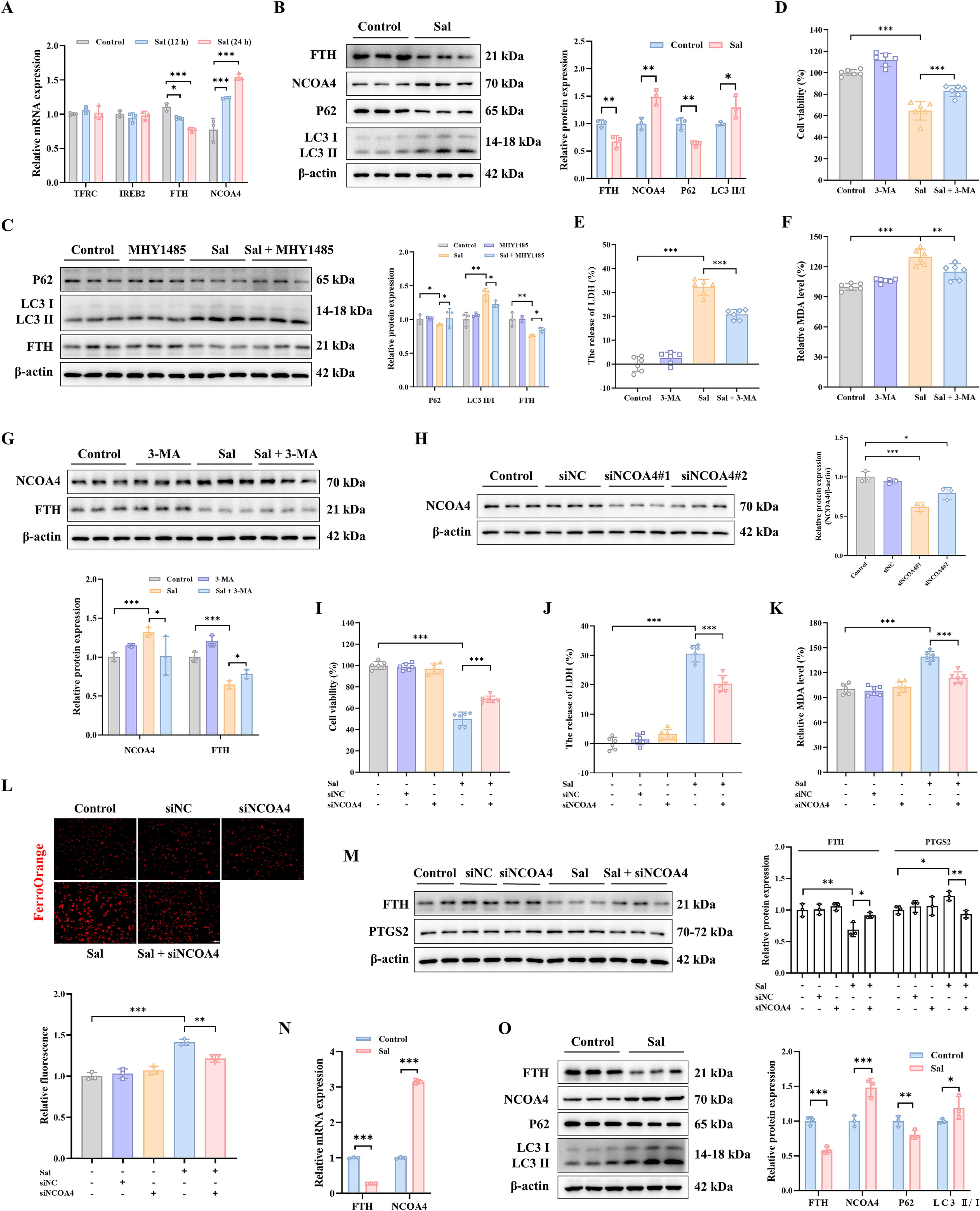
为了进一步探索三阴性乳腺癌中自噬相关通路与铁死亡之间的关联,检测了Sal处理后mRNA水平上铁调节蛋白的变化,如转铁蛋白受体(TFRC)、铁反应元件结合蛋白2(IREB2)、铁蛋白重链(FTH)和NCOA4。观察到FTHmRNA下调,NCOA4mRNA显著上调,而其他的没有变化(图8A),这表明Sal诱导的铁死亡可能与铁蛋白自噬相关。随后,我们分析了Sal处理后铁蛋白自噬相关蛋白水平的变化,包括FTH、NCOA4、P62和LC3II/I。如图所示,在Sal处理的MDA-MB-231细胞中,NCOA4和LC3II/I水平上调,而FTH和P62显著下调(图8B)。令人惊讶的是,mTOR激活剂MHY1485显著抑制了Sal增加的LC3II/I水平,并改善了Sal降低的P62和FTH蛋白水平(图8C),这表明Sal激活的铁蛋白自噬可能是通过抑制mTOR通路实现的。
此外,为了研究铁蛋白自噬是否参与了Sal引起的铁死亡,用3-MA预处理细胞以抑制自噬体组装。结果显示,3-MA预处理显著提高了细胞活力,并抑制了Sal诱导的MDA表达(图8D-F)。此外,在Sal组中FTH蛋白水平下调,但在Sal+3-MA组中上调(图8G)。接下来,使用针对NCOA4的siRNA来研究NCOA4是否介导Sal触发的铁蛋白自噬,并通过WB证实转染功效(图8H)。据此,选择siNCOA4#1用于进一步研究。敲低NCOA4使MDA-MB-231细胞抗铁死亡(图8I-J),这由下调的MDA和0ec917b8-67c1-4bf5-8485-1d69dea6857f水平所证明(图8K-L)。与此一致,敲低NCOA4还抑制PTGS2蛋白水平,同时提高Sal诱导的FTH蛋白水平(图8M)。所有结果表明,铁死亡中自噬相关传代是NCOA4介导的铁蛋白自噬,而Sal诱导的铁超载可能归因于由mTOR通路调节的NCOA4介导的铁蛋白自噬。
随后,一项体内实验证实了NCOA4介导的铁蛋白自噬和铁死亡在盐霉素处理中的作用。三阴性乳腺癌(TNBC)。与对照组相比,盐霉素组中LC3II/I和NCOA4的水平升高,而P62和FTH显著降低(图8N-O)。这与体外结果一致,表明盐霉素可能通过mTOR介导的NCOA4调节的铁自噬促进铁死亡的发生。

总结:文章阐明了与铁死亡密切相关的TNBC发病机制,并确定了TNBC中的潜在生物标志物,文章思路非常值得学习。傲星生物有丰富的分析方案、完善的下游验证、机制研究服务,一对一专属服务为您排忧解难,助您轻松应对毕业和晋升!

