


代谢组+网络药理学+分子动力学模拟,鉴定紫苏叶中对抗高尿酸血症的抑制剂!
题目:通过代谢组学分析、网络药理学和分子动力学模拟鉴定紫苏叶中对抗高尿酸血症的抑制剂
英文名:Identification of inhibitors from a functional food-based plant Perillae Folium against hyperuricemia via metabolomics profiling, network pharmacology and all-atom molecular dynamics simulations
杂志:frontiers in Endocrinology
影响因子:3.9
发表时间:2024年2月16日
研究背景:高尿酸血症(HUA)是一种由嘌呤代谢功能障碍引起的代谢紊乱,其中嘌呤水平升高可部分归因于海鲜消费。紫苏叶(PF)历来被用于缓解海鲜引起的疾病。然而,其对HUA的疗效和潜在机制仍不清楚。
研究思路:进行网络药理学分析以确定PF治疗HUA所涉及的候选靶点和潜在机制。通过基因本体论(GO)和京都基因与基因组百科全书(KEGG)分析预测潜在机制。在AutoDockVina和PyRx中进行分子对接,以预测草药化合物与HUA相关靶点之间的结合亲和力。通过分子动力学(MD)模拟、文本挖掘和非靶向代谢组学分析进一步验证了这些抑制剂在HUA中的作用。
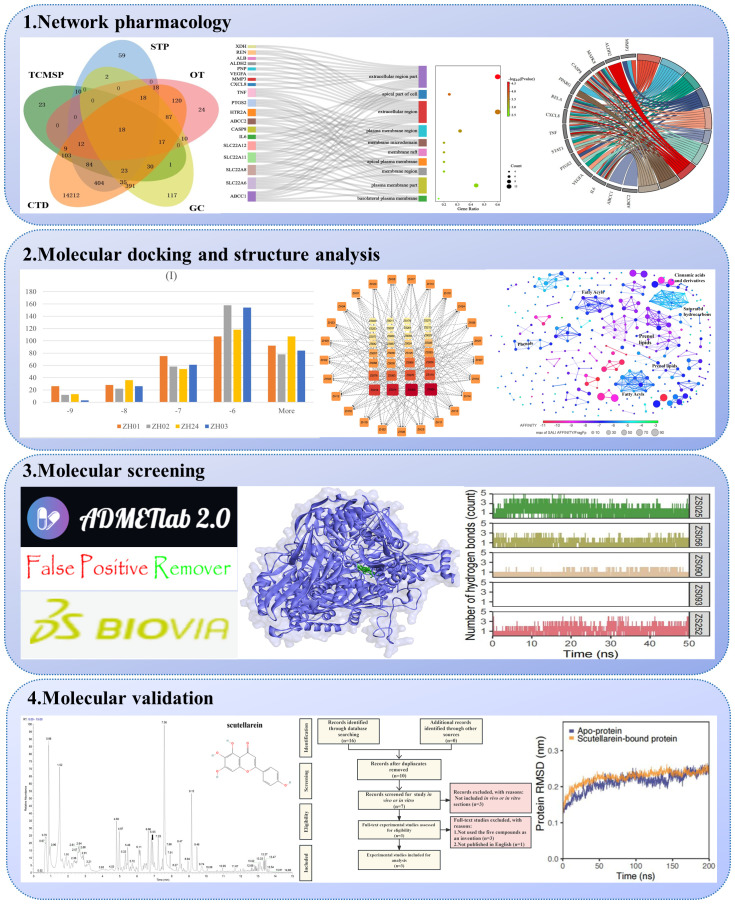
图1
研究结果:
1、紫苏叶治疗高尿酸血症的草药化合物及潜在靶点
通过网络药理学分析,并得到了18个靶点,包括黄嘌呤脱氢酶(XDH)、基质金属蛋白酶-3(MMP3)、单胺氧化酶A型(MAOA)、线粒体醛脱氢酶(ALDH2)、丝裂原活化蛋白激酶8(MAPK8)、Caspase-凋亡酶(Caspase-凋亡酶)8(CASP8)、过氧化物酶体增殖激活受体γ(PPARG)、转录因子p65(RELA)、趋化因子(C-X-C矩阵)配体8(CXCL8)、肿瘤坏死因子(TNF)、转录信号转导和激活因子3(STAT3)、前列腺素G/H合成酶2(PTGS2)、肾素(REN)、雌激素(ESR1)、白蛋白(ALB)、5-羟色胺受体2A(HTR2A)、血管内皮生长因子A(VEGFA)和白细胞介素6(IL6)(图2A)。根据STRING数据库中的25个候选靶点构建了一个PPI网络(图2B)。如图2B所示,这些靶标被分为红色、绿色和蓝色三组。该网络的25个节点之间有102条边,每个节点至少与一个其他节点相关联。
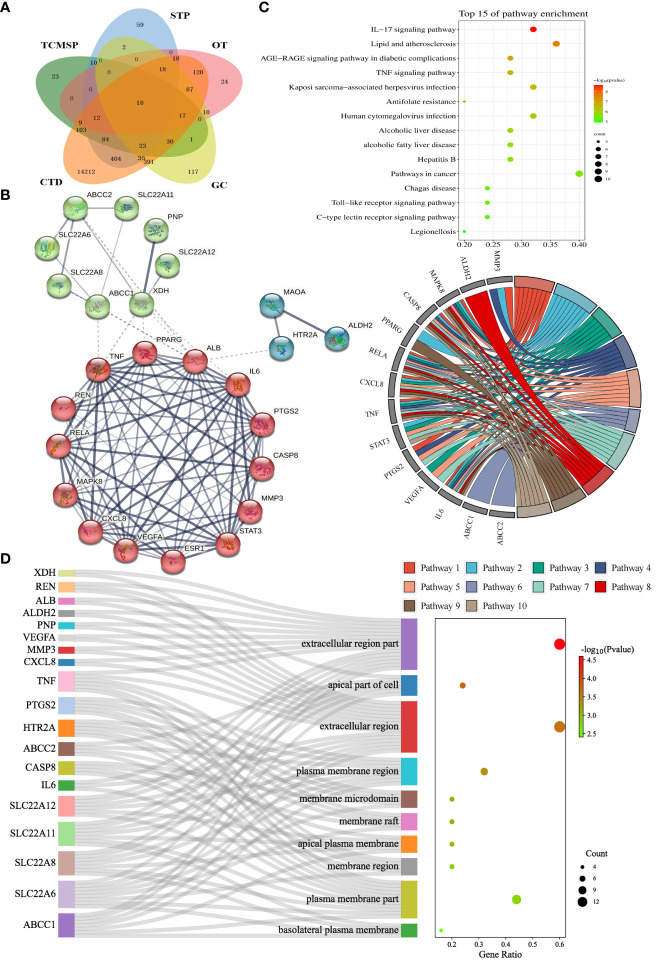
图2
2、紫苏叶参与多种信号传导途径
为阐明化合物-靶标相互作用的可能机制,进行了GO和KEGG富集分析,在15个通路中,癌症通路的基因比例maximum(图2C)。在GO分析中,通过BP、CC和MF分析对25个潜在靶标进行了注释,并根据p值将相应的术语从小到大排序(图2D)。根据CC分析(图2D),细胞外区域部分、细胞顶端部分、细胞外区域、质膜区域、膜筏、膜微域、顶端质膜、膜区域、质膜部分、基底侧和质膜被确定为前十大类别。
3、评估与高尿酸血症蛋白对接的紫苏叶化合物
为了根据相应的分子结构探索PF化合物是否与HUA的潜在靶点存在生物学联系,328种成分与25种HUA靶点进行了对接蛋白质,产生了8200个对接结果,结合得分最好的前10个靶点是ZH01(XDH)、ZH02(MMP3)、ZH24(ABCC1)、ZH03(MAOA)、ZH15(ALB)、ZH23(SLC22A8)、ZH22(SLC22A6)、ZH16(HTR2A)、ZH12(PTGS2)、和ZH05(MAPK8)。其中,XDH的平均结合能minimum(-6.76kcal/mol),表明XDH与328个PF化合物结合良好。在直方图中,根据平均结合亲和力对25个目标物进行了排序,并将其分为四组(图3)。具体来说,(I)组对应的是排名前一至四位的靶标,而其余21个靶标则平均分为三组,即(II)组、(III)组和(IV)组。如图3所示,至少有一种配体与(I)组至(III)组的靶标具有高亲和力(结合分数≤-9kcal/mol),有20多种配体对ZH01(only的一种)具有类似的作用。此外,大多数化合物(超过50%)与(I)组和(II)组目标物的对接亲和力为间歇性(-9至-6kcal/mol)。相反,大多数化合物与(III)类和(IV)类靶标的结合亲和力较弱(>-6kcal/mol),特别是ZH09和ZH17,它们的结合配体数都约为300(91.4%)。
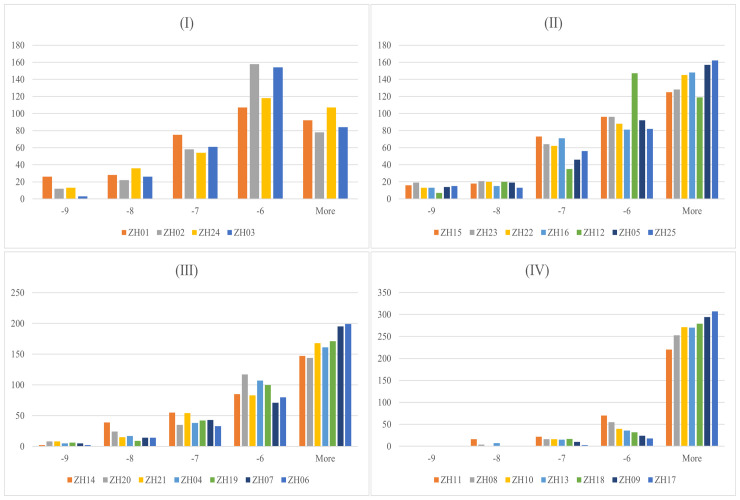

图3
4、化合物-靶标网络揭示了潜在的多靶标多活性化合物机制
根据化合物的结合能,选出了每个靶标的前10个活性化合物。剔除重复的化合物后,共保留了32个活性化合物来构建网络(图4)。在32个活性化合物中,有24个与一个以上的靶点表现出良好的结合能力。有10种化合物与25个靶点中一半以上的靶点结合良好。值得注意的是,ZS059和ZS058的度值均为21。上述发现可能凸显了PF对HUA的协同作用,其特点是多靶点和多组分机制。

图4
5、根据328种化合物的结构及其亲和力进行聚类分析至XDH
随后对328种PF化合物进行了基于结构的分析。如图5a所示,大多数PF化合物与这两种抑制剂的相似性较弱。在图5B中,328种化合物根据其相应的结合能以不同颜色显示为节点。值得注意的是,maximum聚类中的节点往往呈紫红色,这表明它们通常具有良好的结合亲和力(图5B)。
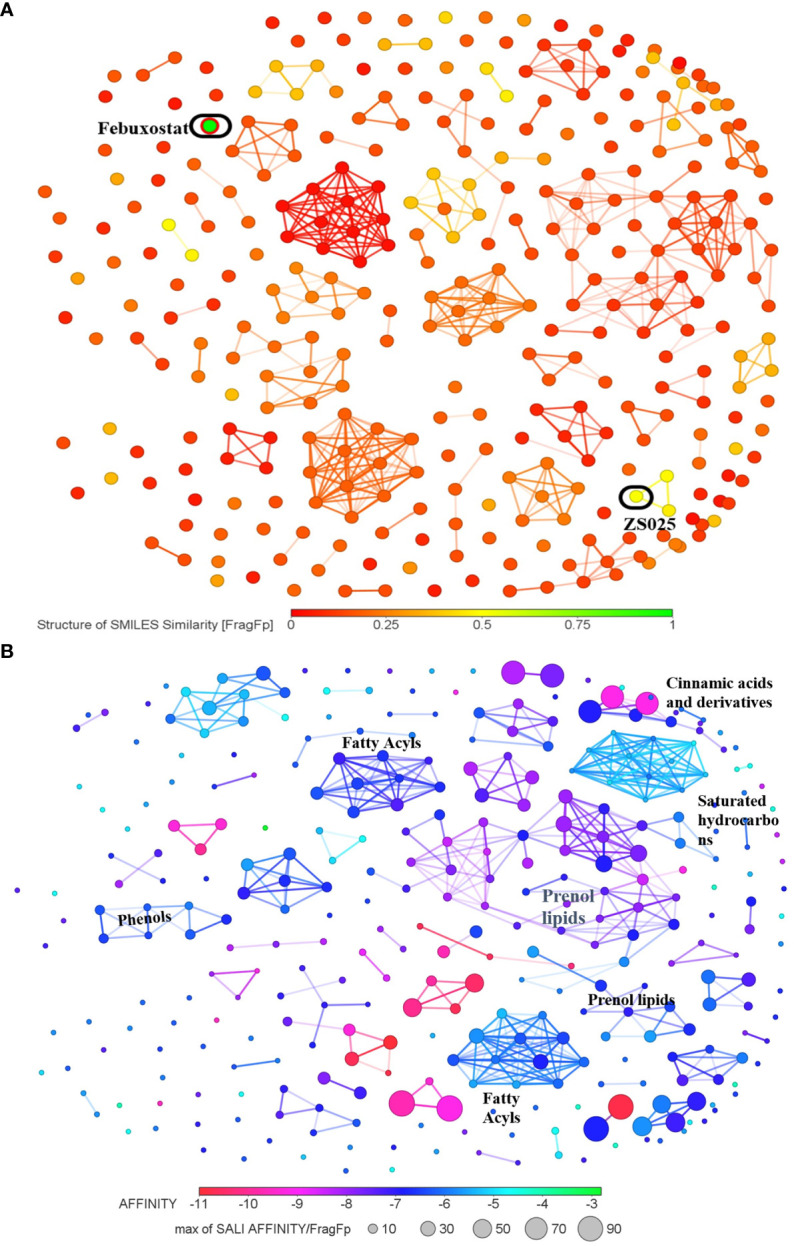
图5
6、五种与XDH有强烈相互作用的配体具有类似于药物的性质,并且非有毒分子
为了进一步探索哪些成分更有可能成为HUA的潜在抑制剂,考虑了一些参数,包括泛检测干扰化合物(PAINS)、氢键(H-bonds)、结合亲和力和ADMET特性。经过筛选,确定了5个合格化合物,包括ZS025、ZS056、ZS090、ZS093和ZS252。如图6所示,ZS090与XDH形成了一种而ZS025、ZS056和ZS093则在同一区域形成了两个H键。除了在两个活性位点残基(Gln1195和Arg913)上形成H键外,ZS252还通过不利的供体-供体键与Ser1081相互作用,距离分别为2.25a、2.85a和1.14Å。
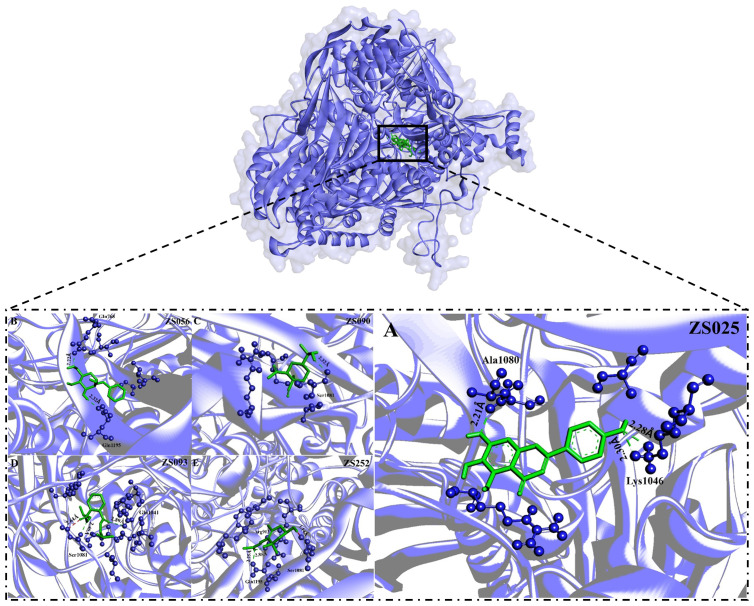
图6
7、分子动力学模拟五种潜在抑制剂
如图7所示,利用结构和动力学量(包括RMSD、RMSF、Rg、SASA和氢键数量)分析了六个模拟系统的构象稳定性和灵活性。如图7A所示,蛋白质-配体复合物在系统弛豫后约10ns达到平衡,这一点从曲线在此时间后形成高原可以看出。这表明ZS025具有更高的稳定性、与XDH活性位点上的其他配体相比,它在很大程度上保留了其初始结构(图7B)。对于Rg和SASA,用Rg测得的蛋白质构象随时间的变化见图7C,用SASA测得的变化见图7D。如图7E和F所示,RMSF和RMSF计算的变化主要集中在Thr750至Val1333之间的结合位点残基上。大部分峰值都位于环状区域周围,并且相互高度重叠。此外,波谷也呈现出类似的趋势,尤其是配体结合型结构。

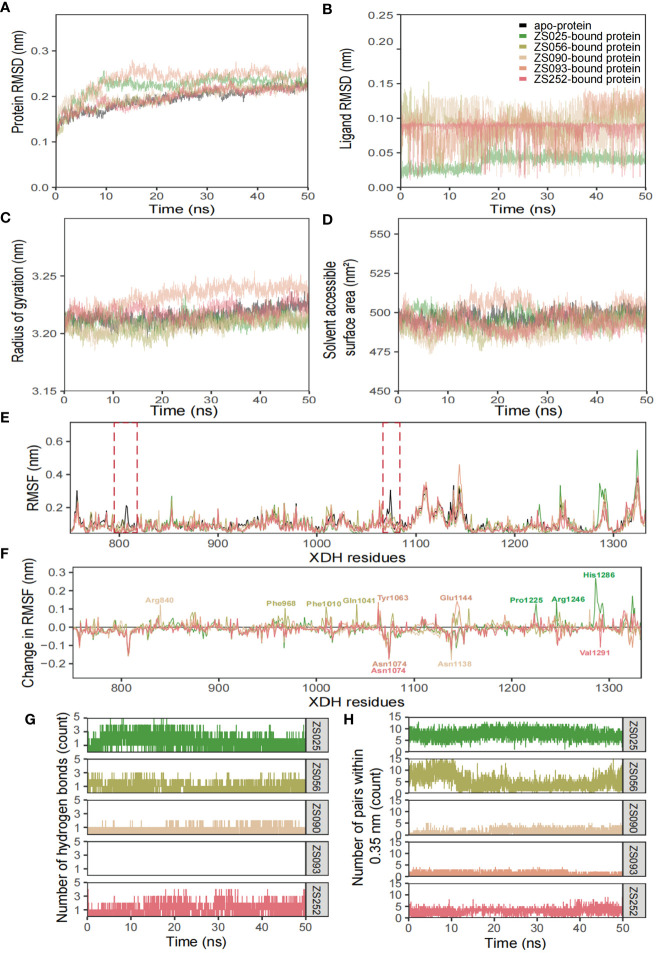
图7
8、根据文献挖掘和非靶向代谢组学研究,证明黄芩苷是一种潜在的XDH抑制剂
为了找到这五种化合物在体外和体内对尿酸产生生物效应的实验室证据,在PubMed、Scopus和CINAHL的基础上进行了文本挖掘分析。搜索的记录来自最早的可用记录至2023年6月28日。查询框中添加的关键词包括黄芩苷、苄基alpha-mannopyranoside、邻苯二甲酸二异丁酯、榄香素、邻苯二甲酸二异丁酯、(3R)-羟基-beta-酮、高尿酸血症、尿酸、黄嘌呤氧化酶和黄嘌呤脱氢酶。考虑了所有可用的关于这五种潜在化合物的英文体内和体外实验研究。剔除重复结果后,剩下10条记录可供进一步分析。四篇已发表的文章中都报道了黄芩苷。这些论文的结果主要基于体外研究,所进行的研究主要是黄嘌呤氧化酶抑制活性测定。然而,除黄芩苷外,其余四种化合物在作者的鉴定研究中未见报道。此外,作者还比较了这五种抑制剂与非布索坦的结构,结果显示ZS025的相似度highest,为0.50877,呈浅黄色。这一发现也与上述结果(图5a)一致。基于上述发现,进一步研究中重点关注了ZS025。随后,为了证实ZS025存在PF颗粒的混合物中,使用UPLC-ESI-Q-Orbitrap-MS进行了非靶向代谢组学分析。在质谱结果中发现了543个分子。在这543个分子中,有22个与从中医药信息平台数据库中检索到的分子相吻合,如迷迭香酸、黄芩苷、鱼腥草素、木犀草素7-O-beta-D-葡萄糖苷和木犀草素。
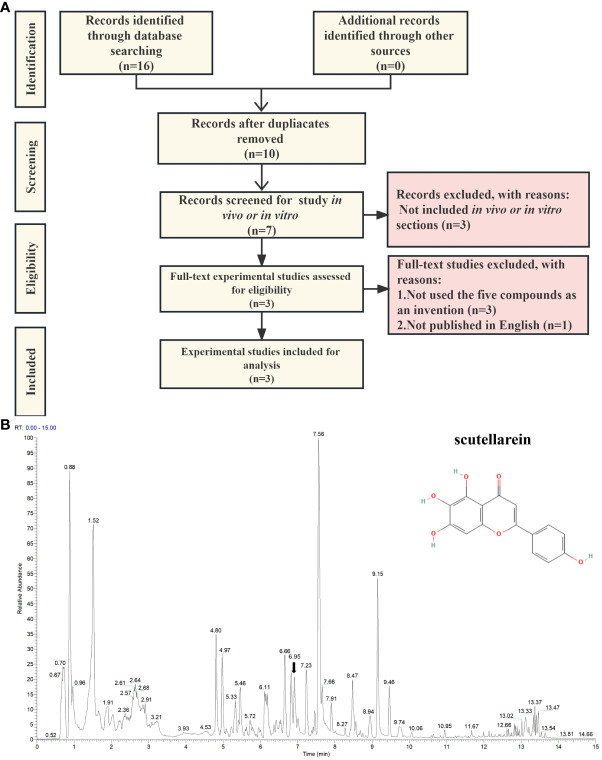
图8
9、黄芩苷-XDH复合物200ns的分子动力学模拟
图9A-F描述了时间序列分析结果比较了apoXDH蛋白和黄芩苷结合的XDH蛋白。黄芩素在大约20ns的平衡后保持了相似的RMSD、Rg和SASA值,平均值分别为0.213nm、3.21nm和489.2Å2。这些结果与apo-XDH蛋白的结果差别不大:RMSD为0.218nm,Rg为3.22nm,SASA为489.5Å2。从图9A-C中的RMSD、Rg和SASA值可以看出,与没有配体的情况相比,在这个较长的模拟过程中,与黄芩素结合的蛋白质折叠更紧凑、更稳定。此外,随着模拟时间的延长,氢键频率的增加也反映了进一步紧密结合的可能性。最初,平均1-2个氢键可稳定黄芩素的结合。大约75ns后,氢键增加到3-4个,如图9E中的高密度粗带所示。虽然图9F中0.35nm范围内的键对数量在系统弛豫20ns后不久略有下降,但波动的幅度和程度保持一致,呈正弦波趋势。如图9H所示,黄芩苷(橙色)与XDH(蓝色)结合的快照从模拟的初始阶段到最后阶段都显示配体与XDH活性位点紧密结合。图9G中的每个残基RMSF图也支持时间序列分析,表明与黄芩素结合后XDH活性位点残基的稳定性更高,波动更小。

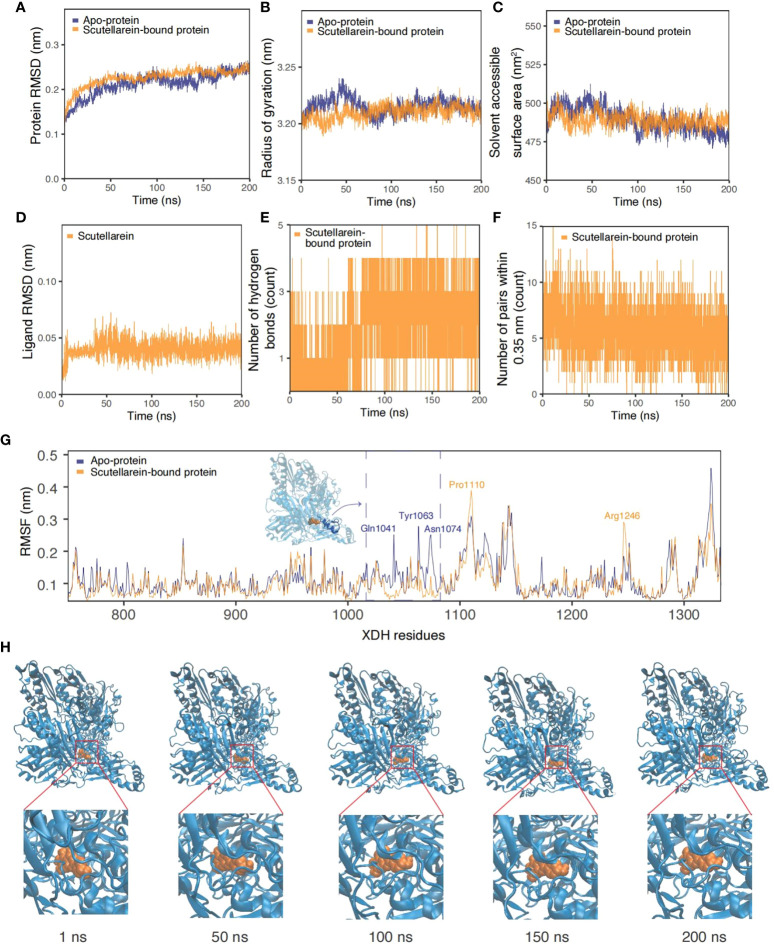
图9
总结:本研究通过网络药理学分析确定PF治疗HUA所涉及的候选靶点和潜在机制,并在分子水平上充分阐明了黄芩苷作为黄嘌呤脱氢酶抑制剂的巨大潜力,为未来的药物设计和开发带来了希望。

